
More Information
Submitted: February 24, 2021 | Approved: March 11, 2021 | Published: March 12, 2021
How to cite this article: Bahador D, Mohammadi A, Foroughi A, Alirezaie B. Sequence-independent single-primer-amplification (SISPA) as a screening technique for detecting unexpected RNA viral adventitious agents in cell cultures. Arch Biotechnol Biomed. 2021; 5: 008-012.
DOI: 10.29328/journal.abb.1001022
Copyright License: © 2021 Bahador D, et al. This is an open access article distributed under the Creative Commons Attribution License, which permits unrestricted use, distribution, and reproduction in any medium, provided the original work is properly cited.
Keywords: Adventitious agents; Contamination; Random amplification; RT-PCR

Sequence-independent single-primer-amplification (SISPA) as a screening technique for detecting unexpected RNA viral adventitious agents in cell cultures
Davood Bahador, Ashraf Mohammadi, Abolhasan Foroughi and Behnam Alirezaie*
Department of Human Viral Vaccines, Razi Vaccine and Serum Research Institute (RVSRI), Agricultural Research, Education, and Extension Organization (AREEO), Karaj, Iran
*Address for Correspondence: Behnam Alirezaie, PhD, Department of Human Viral Vaccines, Razi Vaccine and Serum Research Institute (RVSRI), Agricultural Research, Education, and Extension Organization (AREEO), Karaj, Iran, Tel: 98 (26) 34570038. Email: [email protected]; [email protected]
The sequence-independent, single-primer amplification (SISPA) enables the random amplification of nucleic acids, allowing the detection and genome sequencing of different viral agents. This feature of SISPA method provides evidence for application of it in monitoring the presence of adventitious RNA viruses in cell cultures. We evaluated SISPA method for the detection of a challenge RNA virus representing adventitious agent in cell cultures. Besides, by optimizing the SISPA method in our laboratory, we found false-positive results on negative control lanes in electrophoresis gels. To investigate the sources of contamination, false-positive results of SISPA were cloned into Escherichia coli cells, sequenced, and phylogenetically analyzed. This data revealed that the SISPA method can be used as an adjunct method to confirm the absence of unexpected adventitious RNA viruses in cell cultures. The phylogenetic analysis of SISPA contaminant sequences showed that the false-positive results were caused by nucleic acid amplification of commercial cDNA synthesis kit reagents, probably tracing back to expression plasmids and host ribosomal sequences, used for the production of enzymes. Therefore, laboratories using random amplification methods must be constantly aware of the potentials of such contaminations, yielding false-positive results and background noise in the final NGS reads.
Currently many live and inactivated viral vaccines are produced from viruses propagated in different cell substrates, ranging from primary cultures to continuous cell lines. As a consequence, it increases the risk of adventitious virus contamination of the final product. Some of the most common viral contaminations seen in cell cultures include contaminations with various RNA viruses from variety of sources such as animal derived raw materials and laboratory personnel. The impact of such contaminations can be significant for the diagnostic and manufacturing labs [1]. So, assessment of cell culture and confirming the absence of any adventitious agent is one of the most important safety tests during the manufacturing process [2]. The routine testing method for detecting of viral adventitious agents is based on the observation of cytopathic effects (CPEs), hemadsorption of erythrocytes, or immunofluorescence on the indicator cells. In addition, specific tests such as polymerase chain reaction (PCR) and antibody detecting assay are other strategies for detecting known viral adventitious agents. Despite all the strategies for viral adventitious agents testing, there are still great challenges from an industrial perspective due to the fact that biological products can be contaminated with an unexpected noncytopathic viral agent. Accordingly, there is a need for novel methods for screening and detecting unexpected viral adventitious agents. The use of random amplification methods may make it possible to detect unexpected adventitious agents that we were unable to detect before. Sequence-independent, single-primer amplification (SISPA), as a random amplification method, was first developed in 1991 [3]. This method has been used to detect known and/or unknown pathogens in many diagnostic laboratories [4-7]. The random amplification techniques have also been combined with next generation sequencing (NGS) for genome sequencing of viruses, particularly in the case of samples with very small amounts of genomes [8-12].
The aim of this study was screening for detection of adventitious RNA viruses in cell cultures especially in the case of samples that have tested negative for all expected viral adventitious agents. In this study SISPA method was performed for the detection of an infective challenge RNA virus representing adventitious agent in cell culture supernatant. In addition, by optimization of the SISPA method in our laboratory, we found false-positive results on negative control lanes in electrophoresis gels. To investigate the sources of contaminants, all reagents, water, disposables, and instruments were replaced and renewed. Also, to confirm the source of contamination, false-positive results of SISPA were cloned into Escherichia coli cells, sequenced, and phylogenetically analyzed.
Sequence independent PCR amplification
In this study, the SISPA method was performed as previously described [7]. Briefly, particle-associated nucleic acids were purified from the cell debris, bacteria, and free nucleic acids (DNAs and RNAs) by filtration through a 0.2-µM filter (Sartorius, Germany). Also, nuclease treatment was performed through digestion with a cocktail of DNase (TURBOTM DNase, Ambion, USA), Benzonase (Sigma-Aldrich, Denmark), and RNase (Bio Basic Inc., Canada).
Viral genomes were extracted from samples, using FavorPrepTM Viral Nucleic Acid Extraction Kit (Favorgen, Taiwan). Next, first-strand cDNA was synthesized in a 20-µL reaction mixture, containing 12 µL of viral nucleic acids, 100 pmol of primer K-8N (GAC CAT CTA GCG ACC TCC CAN NNN NNN N), 200 U of moloney murine leukemia virus (MMuLV) RT (YTA, Iran), and 0.5 mM of each dNTP solution. Afterward, 20 µL of cDNA was heated to 70 °C for five minutes and cooled down to 4 °C in the presence of 20 pmol of primer K-8N in 1X Klenow buffer. In order to produce double-stranded DNAs with the fixed portion of primer K-8N from generated first stranded cDNAs, 5 U of Klenow fragment (Takara Bio, Japan) was added and incubated at 37 °C for 60 minutes. The synthesized cDNA was subsequently used for the polymerase chain reaction (PCR) assay.
The PCR amplification was carried out in a 50-µL mixture, containing 10 µL of first-strand cDNA product, 1X PCR buffer, 0.8 µM of primer K (GAC CAT CTA GCG ACC TCC AC), and 2.5 U of Taq DNA polymerase (YTA, Iran). The initial denaturation step was performed at 95 °C for five minutes, followed by five cycles of denaturation for one minute at 95 °C, primer annealing for one minute at 59 °C, and elongation for one minute at 72 °C. Thirty-three more cycles were also performed as follows: 95 °C for 20 seconds, 59 °C for 20 seconds, 72 °C for one minute +2 seconds for each cycle. The final elongation step was performed at 72 °C for seven minutes.
Cloning and sequencing
The DNAs resulting from SISPA method were ligated into pTG19-T plasmids (PCR TA Cloning Kit, Sinaclon, Iran) following purification on 1% agarose gel. The E. coli XL1-blue strain was transformed into recombinant plasmids, according to the manufacturer’s instructions. Colonies containing recombinant plasmids were screened by blue-white screening. Plasmids were extracted using FavorPrepTM Plasmid Extraction Mini Kit (Favorgen, Taiwan). The sizes of plasmids were checked on agarose gel. Extracted plasmids were also screened for the presence of inserts, using PCR with universal M13 primers and the K primer. PCR with M13 forward and backward primers was performed according to the manufacturer’s instructions (PCR TA Cloning Kit, Sinaclon, Iran). The PCR with K primer was carried out by adding 1 µL of plasmid in 24 µL of PCR cocktail consisting of 1 X PCR buffer, 0.04 mM of each dNTP, 2.5 U of Taq DNA polymerase (YTA, Iran), and 0.8 µM of K primer (as mentioned earlier). Cycling parameters were programed as initial denaturation at 95 °C for 5 minutes followed by 33 cycles of denaturation at 95 °C for 1 minute, annealing at 59 °C for 1 minute, and elongation at 72 °C for 1 minute, followed by final elongation at 72 °C for 7 minutes. The PCR amplicons were checked on 1% agarose gels. Then, sequencing was performed in both directions, using universal M13 forward and reverse primers to ensure the quality of data.
Limit of detection
For estimate of test’s limit of detection, extracts of the dilution series of mumps virus (Hoshino strain) was used. The history of the development and of attenuated mumps virus Hoshino vaccine strain has been described previously [13,14]. In the present study, this strain was available in-house. The current stock was propagated in chicken embryonic fibrobalsts (CEFs) and extracted from the cell debris through a 0.2-µm syringe filter (Sartorius, Germany). Suspension containing approximately 106 CCID 50/ml of virus was serially diluted in sterile DEPC-treated water. RNAs were extracted from the samples and RT-PCR amplification was performed as previously described. After staining the gel, the last visible DNA band was extracted, cloned and sequenced as previously described.
Contamination detection
To confirm the source of contamination, false-positive results of SISPA were also cloned into Escherichia coli cells and sequenced as previously described.
Data analysis
The homology of sequences with the GenBank-deposited sequences was searched, using the Basic Local Alignment Search Tool (BLAST). DNAMAN software package (version 4.13) was also used for phylogenetic studies. Phylogenetic trees were constructed from DNA sequences, using the neighboring-joining method. Also, the bootstrap test was applied 1000 times.
Detection of challenge virus representing an adventitious agent and limit of detection
To determine whether this method detect adventitious RNA viruses, optimized method was tested to detect challenge virus representing an adventitious agent using virus-containing supernatants (Hoshino strain of mumps virus). Mumps virus is an enveloped single-stranded negative-sense RNA virus which some chick and mammalian cells are permissive to its replication.
It is important that the minimum viral copy number that is able to show visible and clear smear in the SISPA method to be determined. To accomplish this purpose, 10-fold serial dilutions of mumps virus with known titer were prepared. Regarding to the results, the lowest detectable viral copy number was considered as 102 mumps viral particles per RT-PCR reaction (Figure 1A). Afterwards, fragments resulting from each dilution were cloned into plasmids. In order to screening the presence of inserts in plasmids, PCR with universal M13 primers and the K primer were carried out. The PCR amplicons were checked on agarose gel, and the inserts producing two sharp bands were considered to contain inserts flanked by M13 and K primer sequences (Figure 1B and 1C). Thereafter, several positive clones were used for sequencing. The sequencing results showed that all sequences were flanked by K primer sequences at both ends. The sequences from supernatants of mumps virus showed the highest level of homology (100%) to previously sequenced genomes of this strain available at GenBank (accession numbers AB470486 and MK279727). The results also revealed that although 102 viral particles was able to show visible smear in agarose gel, 105 particles seemed to result in sufficient amount of specific viral PCR product. In other words, in the case of samples that contain viral copy number below 105 particles, most clones contained non-specific sequences. So, the results showed that the SISPA method is efficient as a method for detecting of adventitious agents for samples with greater than 105 viral particles per reaction. These conditions seem to be ideal for screening of adventitious viral agents in cell cultures because most samples contain more of viral copy number as a result of enrichment of detectable progeny virus populations by propagation in permissive cell substrates.
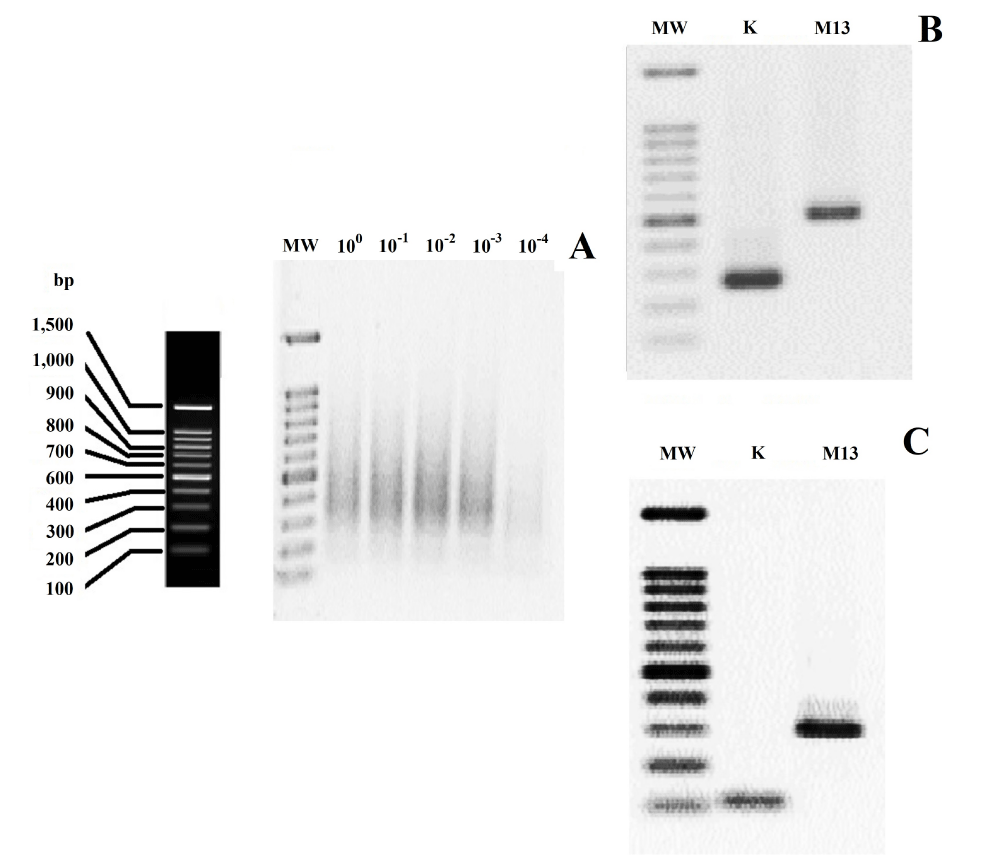
Figure 1: (A) Estimation of the detection limit of SISPA method using 10-fold serial dilution of the Challenge Virus Representing an Adventitious Agent. The mumps virus containing 106 CCID50/ml was serially diluted and each dilution subjected to method. (B and C) Agarose gel electrophoresis of PCR amplicons of the recombinant plasmids with universal M13 primers and the K primer (two of the several reactions are shown here). Lane MW is the 100 bp DNA lader marker (YTA, Iran).
Contaminant sequences
False-positive results (clear smears in size range of 50-800 bp with few prominent bands) were obtained following the application of the SISPA method. It was suspected that the amplification reagents were the source of contamination. This could be associated with the presence of trace amounts of nucleic acids (e.g., expression vectors and prokaryotic expression host compounds), which are commonly used for the production of commercial enzymes. To approve this assumption, false-positive results of SISPA were cloned into E. coli cells, sequenced for both strands, and phylogenetically analyzed.
Several clones, which were sequenced, showed the following homologies. All sequences were flanked by K primer sequences at both ends. Some clones carried murine leukemia virus sequence. The standard nucleotide BLAST analysis revealed that the sequences had the highest level of homology (100%) with several sequences of the M-MuLV pol gene (Figure 2A). The sequencing of other amplicons showed high homology (100%) with BL21 E. coli 23s rRNAs. High sequence homology was also found with other strains of E. coli rRNAs (Figure 2B,C). Detection of M-MuLV pol gene and E. coli rRNA sequences in false-positive result amplicons showed that residual nucleic acid sequences in commercial cDNA synthesis reagents were the cause of false-positive results. The only realistic explanation that accounts for these observations is that cDNA synthesis reagents contained trace amounts of nucleic acids. The presence of murine leukemia virus (MLV) sequences in other commercial laboratory reagents have been previously reported using RT-PCR assay, with primers specific for xenotropic murine leukemia virus-related virus (XMRV) and MLV [15-17]. Previous studies have also reported contamination of some other commercial reagents with different bacterial genomes and other DNAs [18-24]. The present study showed that the trace amounts of nucleic acids in commercial laboratory reagents, used in random amplification methods, may be problematic, yielding false-positive results and background noise in the final NGS reads.
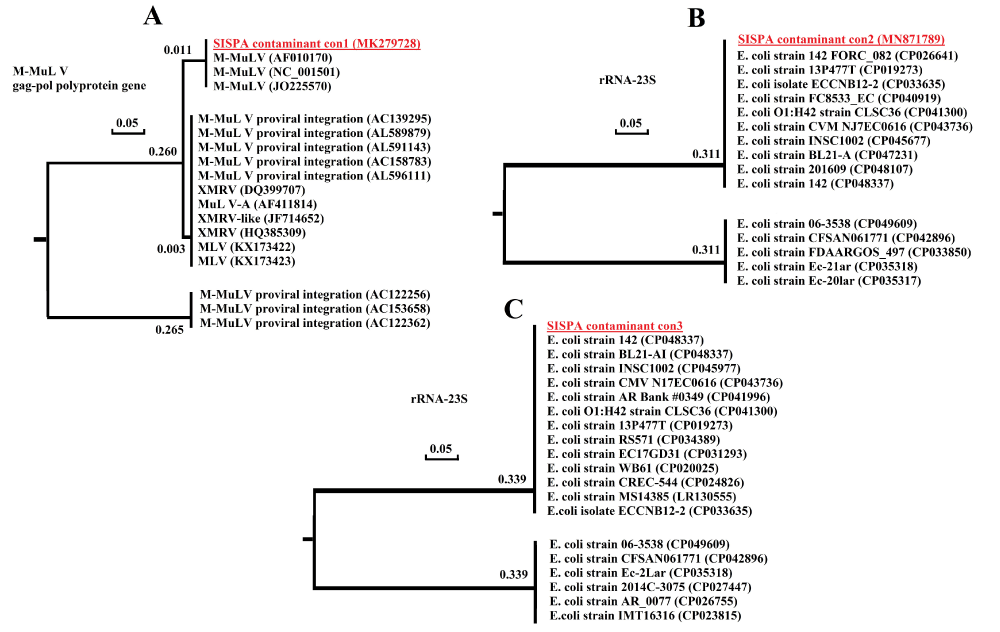
Figure 2: Phylogenetic relationship between SISPA contaminant sequences (three clones of several different clones is shown here) and GenBank-deposited sequences (high homology). The GenBank accession numbers are shown in parentheses, and the contaminant sequences are bolded and underlined. Branch lengths are shown for each branch point.
In conclusion, SISPA method should be particularly useful for screening of cell cultures for unexpected adventitious RNA viruses that have tested negative for all expected viral adventitious agents. The present finding also indicated that without a confirmatory test, a commercial cDNA synthesis kit should not be used for random amplification methods, such as SISPA. Moreover, the results highlighted the need for controlling different steps of random amplification methods to validate their use for diagnostic purposes and NGS. Also, care should be taken regarding the contamination of other SISPA regents.
The authors thank personnel of human viral vaccine department of Razi vaccine and serum research institute for their technical assistance.
- Petriccani J, Sheets R, Griffiths E, Knezevic I. Adventitious agents in viral vaccines: lessons learned from 4 case studies. Biologicals. 2014; 42: 223-236. PubMed: https://pubmed.ncbi.nlm.nih.gov/25135887/
- Cleveland MH, Anekella B, Brewer M, Chin PJ, Couch H, et al. Report of the 2019 NIST-FDA workshop on standards for next generation sequencing detection of viral adventitious agents in biologics and biomanufacturing. Biologicals. 2019; 64: 76-82.PubMed: https://pubmed.ncbi.nlm.nih.gov/32094072/
- Reyes GR, Kim JP. Sequence-independent, single-primer amplification (SISPA) of complex DNA populations. Molecular and Cellular Probes. 1991; 5: 473-481. PubMed: https://pubmed.ncbi.nlm.nih.gov/1664049/
- Ambrose HE, Clewley JP. Virus discovery by sequence‐independent genome amplification. Rev Med Virol. 2006; 16: 365-383. PubMed: https://pubmed.ncbi.nlm.nih.gov/16929467/
- Bexfield N, Kellam P. Metagenomics and the molecular identification of novel viruses. Veterin J. 2011; 190: 191-198. PubMed: https://pubmed.ncbi.nlm.nih.gov/21111643/
- Djikeng A, Halpin R, Kuzmickas R, Depasse J, Feldblyum, Sengamalay N, et al. Viral genome sequencing by random priming methods. BMC genomics. 2008; 9: 1-9. PubMed: https://pubmed.ncbi.nlm.nih.gov/18179705/
- Victoria JG, Kapoor A, Dupuis K, Schurr DP, Delwart EL. Rapid identification of known and new RNA viruses from animal tissues. PLoS Pathogens. 2008; 4: e1000163. PubMed: https://pubmed.ncbi.nlm.nih.gov/18818738/
- Afonso CL. Sequencing of avian influenza virus genomes following random amplification. Biotechniques. 2007; 43: 188-192. PubMed: https://pubmed.ncbi.nlm.nih.gov/17824386/
- Bishop-Lilly KA, Turell MJ, Willner KM, Butani A, Nolan NM, et al. Arbovirus detection in insect vectors by rapid, high-throughput pyrosequencing. PLoS Neglected Tropical Diseases. 2010; 4: e878. PubMed: https://pubmed.ncbi.nlm.nih.gov/21085471/
- Blomström AL, Widén F, Hammer AS, Belák S, Berg M. Detection of a novel astrovirus in brain tissue of mink suffering from shaking mink syndrome by use of viral metagenomics. J Clin Microbiol. 2010; 48: 4392-4396. PubMed: https://pubmed.ncbi.nlm.nih.gov/20926705/
- Marcacci M, Deluca E, Zaccaria G, Ditommaso M, Mangone I, et al. Genome characterization of feline morbillivirus from Italy. J Virologi Meth. 2016; 234, 160-163. PubMed: https://pubmed.ncbi.nlm.nih.gov/27155238/
- Victoria JG, Kapoor A, Li L, Blinkova O, Slikas B, et al. Metagenomic analyses of viruses in stool samples from children with acute flaccid paralysis. J Virol. 2009; 83: 4642-4651. PubMed: https://pubmed.ncbi.nlm.nih.gov/19211756/
- Sasaki K, Higashihara M, Inoue K, Igarashi Y, Makino S. Studies on the development of a live attenuated mumps virus vaccine. I. Attenuation of the Hoshino “wold” strain of mumps virus. Kitasato Arch Expe Med. 1976; 49: 43-52. PubMed: https://pubmed.ncbi.nlm.nih.gov/828947/
- Makino S, Yammane Y, Sasaki K, Nagashima T, Higashihara M. Studies on the development of a live attenuated mumps virus vaccine. II. Development and evaluation of the live “Hoshino” mumps vaccine. Kitasato Arch Exp Med. 1976; 49: 53-62. PubMed: https://pubmed.ncbi.nlm.nih.gov/1025341/
- Knox K, Carrigan D, Simmons G, Teque F, Zhou Y, et al. No evidence of murine-like gammaretroviruses in CFS patients previously identified as XMRV-infected. Science. 2011; 333L 94-97. PubMed: https://pubmed.ncbi.nlm.nih.gov/21628393/
- Sato E, Furuta RA, Miyazawa T. An endogenous murine leukemia viral genome contaminant in a commercial RT-PCR kit is amplified using standard primers for XMRV. Retrovirology. 2010; 7: 110. PubMed: https://pubmed.ncbi.nlm.nih.gov/21171978/
- Zheng H, Jia H, Shankar A, Heneine W, Switzer WM. Detection of murine leukemia virus or mouse DNA in commercial RT-PCR reagents and human DNAs. PLoS One. 2011; 6: e29050. PubMed: https://pubmed.ncbi.nlm.nih.gov/22205995/
- Champlot S, Berthelot C, Pruvost M, Bennett EA, Grange T, et al. An efficient multistrategy DNA decontamination procedure of PCR reagents for hypersensitive PCR applications. PloS one. 2010; 5: e13042. PubMed: https://pubmed.ncbi.nlm.nih.gov/20927390/
- Evans GE, Murdoch DR, Anderson TP, Potter HC, George PM, et al. Contamination of Qiagen DNA extraction kits with Legionella DNA. J Clin Microbiol. 2003; 41, 3452-3453. PubMed: https://pubmed.ncbi.nlm.nih.gov/12843121/
- Koncan R, Valverde A, Morosini MI, García-Castillo M, Cantón R, et al. Learning from mistakes: Taq polymerase contaminated with β-lactamase sequences results in false emergence of Streptococcus pneumoniae containing TEM. J Antimicrob Chemother. 2007; 60: 702-703. PubMed: https://pubmed.ncbi.nlm.nih.gov/17597059/
- Mühl H, Kochem AJ, Disqué C, Sakka SG. Activity and DNA contamination of commercial polymerase chain reaction reagents for the universal 16S rDNA real-time polymerase chain reaction detection of bacterial pathogens in blood. Diagn Microbiol Infect Dis. 2010; 66: 41-49. PubMed: https://pubmed.ncbi.nlm.nih.gov/18722072/
- Newsome T, Li BJ, Zou N, Lo SC. Presence of bacterial phage-like DNA sequences in commercial Taq DNA polymerase reagents. J Clin Microbiol. 2004; 42: 2264-2267. PubMed: https://pubmed.ncbi.nlm.nih.gov/15131208/
- Thoendei M, Jeraldo P, Greenwood-Quaintance KE, Yao J, Chia N, et al. Impact of contaminating DNA in whole-genome amplification kits used for metagenomic shotgun sequencing for infection diagnosis. J Clin Microbiol. 2017; 55: 1789-1801. PubMed: https://pubmed.ncbi.nlm.nih.gov/28356418/
- Wally N, Schneider M, Thannesberger J, Kastner M, Bakonyi T, et al. Plasmid DNA contaminant in molecular reagents. Sci Rep. 2019: 9: 1-11. PubMed: https://pubmed.ncbi.nlm.nih.gov/30733546/